[1]:
# useful to autoreload the module without restarting the kernel
%load_ext autoreload
%autoreload 2
[2]:
from mppi import InputFiles as I, Calculators as C, Datasets as D, Utilities as U, Parsers as P
from mppi.Calculators import Tools
from mppi.Datasets import PostProcessing as PP
import matplotlib.pyplot as plt
import os
import numpy as np
[3]:
omp = 1
mpi = 4
Analysis of the electron-phonon coupling¶
We analyze the effects of the electron-phonon coupling in the electronic and optical properties of bulk Si.
This analysis is base on the web page : https://www.yambo-code.eu/wiki/index.php/Electron_Phonon_Coupling
[4]:
run_dir = 'Electron_phonon_Si'
Due to the way in which the ph.x is implemented it is more convenient to run each step of the calculations in a specific folder. So the prefix of all the computations is the same but they are exectuded in differen paths.
SCF and NSCF calculations¶
[5]:
rr = C.RunRules(mpi=mpi,omp_num_threads=omp)
code = C.QeCalculator(rr)
code.global_options()
Initialize a QuantumESPRESSO calculator with scheduler direct
[5]:
{'scheduler': 'direct',
'mpi': 4,
'omp_num_threads': 1,
'executable': 'pw.x',
'skip': True,
'clean_restart': True,
'dry_run': False,
'wait_end_run': True,
'activate_BeeOND': False,
'verbose': True}
First we perform a scf calculation on a 8x8x8 k grid to ensure that the electronic density of the GS is converged.
The convergence threshold is set to 1e-12, this value and k grid are loosend w.r.t. the paramteres used in the original tutorial to redcue computational time.
Note that, following the Yambo tutorial, the geometry of the lattice is set using ibrav=0 with the explicit values of the cell parameters. This is done to avoid possible incompatibility between the PW and Yambo conventions for the FCC lattice.
[6]:
prefix = 'si'
name = 'si.scf'
inp= I.PwInput(file='IO_files/Input_for_electron_phonon_Si_tutorial/si.scf.in')
inp.set_prefix(prefix)
inp.set_pseudo_dir('pseudos',abs_path=True)
#print(inp.convert_string())
[7]:
scf_run_dir = os.path.join(run_dir,'scf')
results = code.run(input=inp,run_dir=scf_run_dir,name=name)
results
Skip the run of si.scf
[7]:
'/home/marco/Applications/MPPI/sphinx_source/tutorials/Electron_phonon_Si/scf/si.save/data-file-schema.xml'
Then we perform a nscf calculation on a smaller 4x4x4 grid with 12 bands.
[8]:
inp.set_nscf(12,force_symmorphic=True,conv_thr=1e-12)
inp.set_kpoints(points=[4,4,4])
#print(inp.convert_string())
[9]:
nscf_run_dir = os.path.join(run_dir,'nscf')
results = code.run(input=inp,run_dir=nscf_run_dir,name=prefix,source_dir=os.path.join(scf_run_dir,prefix+'.save'))
results
Skip the run of si
The folder /home/marco/Applications/MPPI/sphinx_source/tutorials/Electron_phonon_Si/nscf/si.save already exists. Source_dir Electron_phonon_Si/scf/si.save not copied
[9]:
'/home/marco/Applications/MPPI/sphinx_source/tutorials/Electron_phonon_Si/nscf/si.save/data-file-schema.xml'
[10]:
data = P.PwParser(results)
Parse file : /home/marco/Applications/MPPI/sphinx_source/tutorials/Electron_phonon_Si/nscf/si.save/data-file-schema.xml
[11]:
kpoints=-data.kpoints
#kpoints[4] = np.array([-0.375000000,-0.125000000,0.125000000])
kpoints
[11]:
array([[-0. , -0. , -0. ],
[-0.125, -0.125, 0.125],
[ 0.25 , 0.25 , -0.25 ],
[-0.25 , -0. , -0. ],
[ 0.125, 0.375, -0.375],
[-0. , 0.25 , -0.25 ],
[ 0.5 , -0. , -0. ],
[ 0.5 , -0.25 , -0. ]])
Phonon calculations¶
With this calculation we can compute the dynamical matrix elements and the dvscf files, these quantities are the analougous of the GS density for the phonon calculations
[139]:
rr = C.RunRules(mpi=mpi,omp_num_threads=omp)
code_ph = C.QeCalculator(rr,executable='ph.x')
#code_ph.global_options()
Initialize a QuantumESPRESSO calculator with scheduler direct
[16]:
prefix = 'si'
name = 'si.phonon'
inp = I.PhInput()
inp.set_prefix(prefix)
inp['inputph']['fildvscf'] = "'si-dvscf'"
inp['inputph']['fildyn'] = "'si.dyn'"
inp['inputph']['electron_phonon'] = "'dvscf'"
inp['inputph']['epsil'] = '.true.'
inp.set_kpoints(Tools.build_pw_klist(kpoints))
#print(inp.convert_string())
[17]:
ph_run_dir = os.path.join(run_dir,'phonon')
results = code_ph.run(input=inp,run_dir=ph_run_dir,name=name,source_dir=os.path.join(scf_run_dir,prefix+'.save'))
create the run_dir folder : 'Electron_phonon_Si/phonon'
copy source_dir Electron_phonon_Si/scf/si.save in the /home/marco/Applications/MPPI/sphinx_source/tutorials/Electron_phonon_Si/phonon/si.save
run command: mpirun -np 4 ph.x -inp si.phonon.in > si.phonon.log
computation si.phonon is running...
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_UNDERFLOW_FLAG IEEE_DENORMAL
computation si.phonon ended
Once that the si.dyn* files and the dvscf files in the ph0 have been computed we can calculate the electron-phonon matrix elements.
We copy these files in the run_dir of the dvscf computation for the evaluation of the electron-phonon matrix elements.
[18]:
dvscf_run_dir = os.path.join(run_dir,'dvscf')
if not os.path.isdir(dvscf_run_dir) : os.mkdir(dvscf_run_dir)
os.system('cp -r Electron_phonon_Si/phonon/_ph0/ Electron_phonon_Si/phonon/si.dyn* Electron_phonon_Si/dvscf')
os.system('cp -r Electron_phonon_Si/nscf/si.save/ Electron_phonon_Si/dvscf/')
[18]:
0
In this case we use the nscf computations as starting point for the electronic quantities. Note that the calculator uses the option clean_restart=False. otherwise the dyn and ph0 files are erased.
[20]:
prefix = 'si'
name = 'si.dvscf'
inp = I.PhInput()
inp.set_prefix(prefix)
inp['inputph']['fildvscf'] = "'si-dvscf'"
inp['inputph']['fildyn'] = "'si.dyn'"
inp['inputph']['electron_phonon'] = "'yambo'"
inp['inputph']['trans'] = '.false.'
inp.set_kpoints(Tools.build_pw_klist(kpoints))
#print(inp.convert_string())
[21]:
results = code_ph.run(input=inp,run_dir=dvscf_run_dir,name=name,clean_restart=False)
run performed starting from existing results
run command: mpirun -np 4 ph.x -inp si.dvscf.in > si.dvscf.log
computation si.dvscf is running...
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
computation si.dvscf ended
Import in yambo¶
We create the yambo dir using the make_p2y function of the module Tools
[12]:
yambo_dir = 'Electron_phonon_Si/yambo'
[13]:
Tools.make_p2y(yambo_dir=yambo_dir,input_dir='Electron_phonon_Si/nscf/si.save')
Create the folder path Electron_phonon_Si/yambo
Executing command: cd Electron_phonon_Si/yambo; p2y -I ../nscf/si.save
Then a link of the elph_dir in the yambo run_dir
[14]:
elph_dir = 'Electron_phonon_Si/dvscf/elph_dir'
if not os.path.isdir('Electron_phonon_Si/yambo/elph_dir'):
src = os.path.abspath(elph_dir)
dest = os.path.abspath('Electron_phonon_Si/yambo/elph_dir')
os.symlink(src,dest,target_is_directory=True)
and we build the r_setup
[16]:
Tools.build_r_setup(yambo_dir=yambo_dir,yambo_command='yambo_ph -J elph_dir')
Build the r_setup in the yambo_dir path Electron_phonon_Si/yambo
We use ypp_ph to generate the electron-phonon database that can be used by yambo
[17]:
inp = I.YamboInput(args='ypp_ph -g g',folder=yambo_dir)
inp.set_scalar_variables(DBsPATH='elph_dir/')
inp
[17]:
{'args': 'ypp_ph -g g',
'folder': 'Electron_phonon_Si/yambo',
'filename': 'yambo.in',
'arguments': ['gkkp'],
'variables': {'PHfreqF': 'none', 'PHmodeF': 'none', 'DBsPATH': 'elph_dir/'}}
[18]:
rr = C.RunRules(mpi=mpi,omp_num_threads=omp)
code = C.YamboCalculator(rr,executable='ypp_ph')
#code.global_options()
Initialize a Yambo calculator with scheduler direct
[20]:
name= 'GKKP'
code.run(input=inp,name=name,run_dir=yambo_dir)
run command: mpirun -np 4 ypp_ph -F GKKP.in -J GKKP -C GKKP
computation GKKP is running...
computation GKKP ended
There are no o-* files.
Maybe you have performed a computation that does not create any output file or wait_end_run
and/or the dry_run option are active.
Otherwise a possible error has occured during the computation
[20]:
{'output': {},
'report': 'Electron_phonon_Si/yambo/GKKP/r-GKKP_gkkp_gkkp_db',
'dft': 'Electron_phonon_Si/yambo/SAVE/ns.db1',
'elph_gkkp': 'Electron_phonon_Si/yambo/GKKP/ndb.elph_gkkp'}
Quasi-particle band structure¶
[21]:
rr = C.RunRules(mpi=mpi,omp_num_threads=omp)
code = C.YamboCalculator(rr,executable='yambo_ph')
#code.global_options()
Initialize a Yambo calculator with scheduler direct
We include the path of the folder with e-p matrix elements using the -J option. Alternatively we could copy the e-p database in the SAVE folder
[22]:
run_dir = 'Electron_phonon_Si/yambo'
[23]:
inp = I.YamboInput(args='yambo_ph -g n -p fan -c ep -V gen -J GKKP',folder=run_dir)
inp.set_extendOut()
inp.set_array_variables(units='eV',GDamping=0.01)
inp.set_kRange(1,1)
inp
[23]:
{'args': 'yambo_ph -g n -p fan -c ep -V gen -J GKKP',
'folder': 'Electron_phonon_Si/yambo',
'filename': 'yambo.in',
'arguments': ['gw0', 'el_ph_corr', 'ExtendOut'],
'variables': {'Nelectro': [8.0, ''],
'ElecTemp': [0.0, 'eV'],
'BoseTemp': [-1.0, 'eV'],
'OccTresh': [1e-05, ''],
'MEM_tresh': [51200.0, 'Kb'],
'SE_Threads': [0.0, ''],
'GDamping': [0.01, 'eV'],
'RandQpts': [0.0, ''],
'DysSolver': 'n',
'GphBRnge': [[1, 12], ''],
'ElPhModes': [[1, 6], ''],
'QPkrange': [[1, 1, 1, 12], '']}}
[25]:
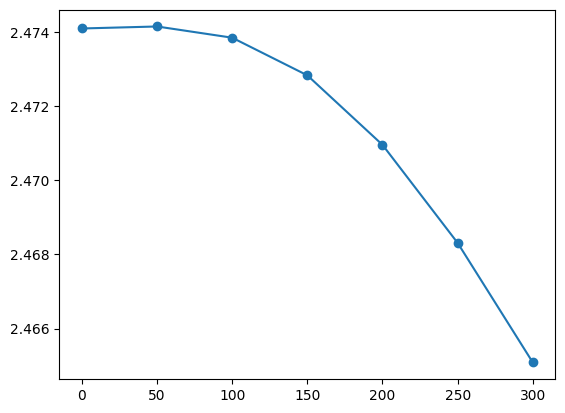
Temp = [0,50,100,150,200,250,300]
study = D.Dataset(run_dir=run_dir,num_tasks=2,verbose=True,skip=True)
study.set_postprocessing_function(PP.yambo_parse_data)
for T in Temp:
inp.set_array_variables(units='K',BoseTemp=T)
idd = 'elph_T%s'%T
study.append_run(id=idd,input=inp,runner=code,jobname =[idd,'GKKP'])
Initialize a Dataset with 2 parallel tasks
[27]:
#study.runs[0]
[29]:
results = study.run()
Run the selection [0, 1, 2, 3, 4, 5, 6] with the parallel task_groups [[0, 1], [2, 3], [4, 5], [6]]
Run the task [0, 1]
Skip the run of elph_T50
delete folder: Electron_phonon_Si/yambo/elph_T0
run command: mpirun -np 4 yambo_ph -F elph_T0.in -J "elph_T0,GKKP" -C elph_T0
computation elph_T0 is running...
computation elph_T0 ended
Task [0, 1] ended
Run the task [2, 3]
Skip the run of elph_T100Skip the run of
elph_T150
Task [2, 3] ended
Run the task [4, 5]
Skip the run of elph_T200
Skip the run of elph_T250
Task [4, 5] ended
Run the task [6]
Skip the run of elph_T300
Task [6] ended
[30]:
gap_G = [results[ind].data.get_gap(k_full=1,band_full=4,verbose=False) for ind in range(len(Temp))]
gap_G
[30]:
[2.474098,
2.4741519999999997,
2.473849,
2.472833,
2.4709570000000003,
2.4683100000000002,
2.465083]
[31]:
plt.plot(Temp,gap_G)
plt.scatter(Temp,gap_G)
[31]:
<matplotlib.collections.PathCollection at 0x7f7a9902fbb0>
[ ]: