[1]:
# useful to autoreload the module without restarting the kernel
%load_ext autoreload
%autoreload 2
[2]:
from mppi import InputFiles as I, Calculators as C, Utilities as U
from mppi.Calculators import Tools
import matplotlib.pyplot as plt
import os
import numpy as np
[3]:
omp = 1
mpi = 4
Analysis of the band structure with QuantumESPRESSO¶
We compute the band structure of Silicon and Gallium arsenide and graphene using the tools of the QuantumESPRESSO and Yambo packages.
[4]:
run_dir = 'Pw_bands'
Band structure of GaAs¶
The first step consists in a scf computation
[5]:
scf_prefix = 'gaas_scf'
nscf_prefix = 'gaas_nscf'
bands_prefix = 'gaas_bands'
[6]:
inp = I.PwInput(file='IO_files/gaas_scf.in')
inp.set_prefix(scf_prefix)
inp.set_energy_cutoff(60)
inp.set_kpoints(points=[6,6,6])
#inp
[7]:
rr = C.RunRules(mpi=mpi,omp_num_threads=omp)
code = C.QeCalculator(rr)
code.global_options()
Initialize a QuantumESPRESSO calculator with scheduler direct
[7]:
{'scheduler': 'direct',
'mpi': 4,
'omp_num_threads': 1,
'executable': 'pw.x',
'skip': True,
'clean_restart': True,
'dry_run': False,
'wait_end_run': True,
'activate_BeeOND': False,
'verbose': True}
[8]:
code.run(input=inp,run_dir=run_dir,name=scf_prefix)
Skip the run of gaas_scf
[8]:
'/home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/gaas_scf.save/data-file-schema.xml'
Before performing the bands computation we make an nscf computation on a regular kpath, this will be useful as input for other tutorials of the package.
[9]:
inp.set_nscf(8,force_symmorphic=True) #use force_symmorphic = True since this computation is used as input of a Yambo computation later
inp.set_prefix(nscf_prefix)
inp.set_kpoints(points=[8,8,8])
#inp
[10]:
code.run(input=inp,run_dir=run_dir,name=nscf_prefix,source_dir=os.path.join(run_dir,scf_prefix)+'.save')
Skip the run of gaas_nscf
The folder /home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/gaas_nscf.save already exists. Source_dir Pw_bands/gaas_scf.save not copied
[10]:
'/home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/gaas_nscf.save/data-file-schema.xml'
Now we perform the bands computation specifying the kpoints on a path.
To define the path we write the coordinates of the high symmetry points (using the tpiba_b type of pw) and we make usage of the function build_kpath
[11]:
G = [0.,0.,0.]
X = [0.,0.,1.]
L = [0.5,0.5,0.5]
W = [1.0,0.5,0.]
K = [0.,1.,1.]
# useful to label the high-sym point on the path
high_sym = {'X':X,'L':L,'G':G,'K':K,'W':W}
The high symmetry points for some lattice structure are also written in the Constants module of the Utilities, for instance
[12]:
U.Constants.high_sym_fcc
[12]:
{'G': [0.0, 0.0, 0.0],
'X': [0.0, 0.0, 1.0],
'L': [0.5, 0.5, 0.5],
'K': [0.0, 1.0, 1.0],
'W': [1.0, 0.5, 0.0]}
[13]:
klist = C.Tools.build_pw_kpath(L,G,X,K,G,numstep=30)
klist
[13]:
[[0.5, 0.5, 0.5, 30],
[0.0, 0.0, 0.0, 30],
[0.0, 0.0, 1.0, 30],
[0.0, 1.0, 1.0, 30],
[0.0, 0.0, 0.0, 0]]
[14]:
inp.set_bands(8)
inp.set_prefix(bands_prefix)
inp.set_kpoints(type='tpiba_b',klist=klist)
#inp
[15]:
results_gaas = code.run(input=inp,run_dir=run_dir,name=bands_prefix,source_dir=os.path.join(run_dir,scf_prefix)+'.save')
results_gaas
Skip the run of gaas_bands
The folder /home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/gaas_bands.save already exists. Source_dir Pw_bands/gaas_scf.save not copied
[15]:
'/home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/gaas_bands.save/data-file-schema.xml'
Once that the computation is over we can create an instance of BandStructure. The gap can be set when we init the class
[16]:
results_gaas
[16]:
'/home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/gaas_bands.save/data-file-schema.xml'
[34]:
bands_gaas = U.BandStructure.from_Pw(results_gaas,high_sym,set_gap=1.42) #,
Apply a scissor of 1.1944603644894034 eV
The class contains some methods that return the bands, the kpath or the position of the high symmetry points on the path
It contains also a plot method that show the band structure
[35]:
%matplotlib inline
plt.title('GaAs bands')
plt.ylim(-8,5)
bands_gaas.plot(plt,selection=[1,2,3,4,5])
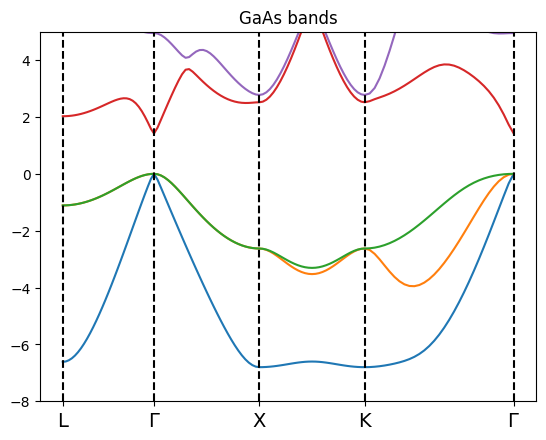
In this case the class build the value of curvilinear abscissa along the paht from the distance among the kpoints on which the QE calculation is performed.
[36]:
bands_gaas.get_kpath()
[36]:
array([0. , 0.02886751, 0.05773503, 0.08660254, 0.11547005,
0.14433757, 0.17320508, 0.20207259, 0.23094011, 0.25980762,
0.28867513, 0.31754265, 0.34641016, 0.37527767, 0.40414519,
0.4330127 , 0.46188022, 0.49074773, 0.51961524, 0.54848276,
0.57735027, 0.60621778, 0.6350853 , 0.66395281, 0.69282032,
0.72168784, 0.75055535, 0.77942286, 0.80829038, 0.83715789,
0.8660254 , 0.89935874, 0.93269207, 0.9660254 , 0.99935874,
1.03269207, 1.0660254 , 1.09935874, 1.13269207, 1.1660254 ,
1.19935874, 1.23269207, 1.2660254 , 1.29935874, 1.33269207,
1.3660254 , 1.39935874, 1.43269207, 1.4660254 , 1.49935874,
1.53269207, 1.5660254 , 1.59935874, 1.63269207, 1.6660254 ,
1.69935874, 1.73269207, 1.7660254 , 1.79935874, 1.83269207,
1.8660254 , 1.89935874, 1.93269207, 1.9660254 , 1.99935874,
2.03269207, 2.0660254 , 2.09935874, 2.13269207, 2.1660254 ,
2.19935874, 2.23269207, 2.2660254 , 2.29935874, 2.33269207,
2.3660254 , 2.39935874, 2.43269207, 2.4660254 , 2.49935874,
2.53269207, 2.5660254 , 2.59935874, 2.63269207, 2.6660254 ,
2.69935874, 2.73269207, 2.7660254 , 2.79935874, 2.83269207,
2.8660254 , 2.91316586, 2.96030631, 3.00744676, 3.05458721,
3.10172766, 3.14886812, 3.19600857, 3.24314902, 3.29028947,
3.33742992, 3.38457038, 3.43171083, 3.47885128, 3.52599173,
3.57313218, 3.62027264, 3.66741309, 3.71455354, 3.76169399,
3.80883445, 3.8559749 , 3.90311535, 3.9502558 , 3.99739625,
4.04453671, 4.09167716, 4.13881761, 4.18595806, 4.23309851,
4.28023897])
Band structure of GaAs with spin-orbit coupling¶
We repeat the computation of the band structure of GaAs including the spin-orbit. To do so we use a full relativistic pseudopotential that is able to include the spin-orbit.
The first step consists in a scf computation
[42]:
scf_prefix = 'gaas_scf_so'
nscf_prefix = 'gaas_nscf_so'
bands_prefix = 'gaas_bands_so'
[43]:
inp = I.PwInput('IO_files/gaas_scf.in')
inp.set_prefix(scf_prefix)
inp.set_energy_cutoff(80)
inp.set_spinorbit()
inp.set_kpoints(points = [6.,6.,6.])
inp.add_atom('Ga','Ga_rel.pz-rrkj3.UPF')
inp.add_atom('As','As_rel.pz-rrkj3.UPF')
inp
[43]:
{'control': {'calculation': "'scf'",
'verbosity': "'high'",
'prefix': "'gaas_scf_so'",
'outdir': "'./'",
'pseudo_dir': "'../pseudos'"},
'system': {'force_symmorphic': '.false.',
'occupations': "'fixed'",
'ibrav': 2,
'celldm(1)': 10.677,
'ntyp': 2,
'nat': 2,
'ecutwfc': 80,
'lspinorb': '.true.',
'noncolin': '.true.'},
'electrons': {'diago_full_acc': '.false.', 'conv_thr': 1e-08},
'ions': {},
'cell': {},
'atomic_species': {'Ga': ['1.0', 'Ga_rel.pz-rrkj3.UPF'],
'As': ['1.0', 'As_rel.pz-rrkj3.UPF']},
'atomic_positions': {'type': 'alat',
'values': [['Ga', [0.0, 0.0, 0.0]], ['As', [0.25, 0.25, 0.25]]]},
'kpoints': {'type': 'automatic',
'values': ([6.0, 6.0, 6.0], [0.0, 0.0, 0.0])},
'cell_parameters': {},
'file': 'IO_files/gaas_scf.in'}
[44]:
rr = C.RunRules(mpi=mpi,omp_num_threads=omp)
code = C.QeCalculator(rr)
Initialize a QuantumESPRESSO calculator with scheduler direct
[45]:
code.run(input=inp,run_dir=run_dir,name=scf_prefix)
Skip the run of gaas_scf_so
[45]:
'/home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/gaas_scf_so.save/data-file-schema.xml'
Before performing the bands computation we make an nscf computation on a regular kpath, this will be useful for the bands analysis using the ypp tools.
We compute 12 bands since in this case the spin degeneracy is equal to 1 and there 8 occupied bands
[46]:
inp.set_nscf(12,force_symmorphic=True) #use force_symmorphic = True since this computation is used as input of a Yambo computation later
inp.set_prefix(nscf_prefix)
#inp
[47]:
code.run(input=inp,run_dir=run_dir,name=nscf_prefix,source_dir=os.path.join(run_dir,scf_prefix)+'.save')
Skip the run of gaas_nscf_so
The folder /home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/gaas_nscf_so.save already exists. Source_dir Pw_bands/gaas_scf_so.save not copied
[47]:
'/home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/gaas_nscf_so.save/data-file-schema.xml'
Now we perform the bands computation specifying the kpoints on a path.
To define the path we write the coordinates of the high symmetry points (using the tpiba_b type of pw) and we make usage of the function build_kpath
[48]:
hsp = U.Constants.high_sym_fcc
[54]:
klist = C.Tools.build_pw_kpath(hsp['L'],hsp['G'],hsp['X'],hsp['K'],hsp['G'],numstep=20)
klist
[54]:
[[0.5, 0.5, 0.5, 20],
[0.0, 0.0, 0.0, 20],
[0.0, 0.0, 1.0, 20],
[0.0, 1.0, 1.0, 20],
[0.0, 0.0, 0.0, 0]]
[57]:
inp.set_bands?
Signature:
inp.set_bands(
nbnd,
conv_thr=1e-08,
diago_full_acc=False,
force_symmorphic=False,
verbosity='high',
)
Docstring:
Set the variables for a bands calculation
Args:
nbnd (:py:class:`int`) : number of bands
conv_thr (:py:class:`float`) : the convergence threshold value
diago_full_acc (:py:class:`bool`)
force_symmorphic (:py:class:`bool`)
verbosity (:py:class:`bool`)
File: ~/Applications/MPPI/mppi/InputFiles/PwInput.py
Type: method
[59]:
inp.set_bands(12,conv_thr=1e-6,force_symmorphic=True)
inp.set_prefix(bands_prefix)
inp.set_kpoints(type='tpiba_b',klist=klist)
inp
[59]:
{'control': {'calculation': "'bands'",
'verbosity': "'high'",
'prefix': "'gaas_bands_so'",
'outdir': "'./'",
'pseudo_dir': "'../pseudos'"},
'system': {'force_symmorphic': '.true.',
'occupations': "'fixed'",
'ibrav': 2,
'celldm(1)': 10.677,
'ntyp': 2,
'nat': 2,
'ecutwfc': 80,
'lspinorb': '.true.',
'noncolin': '.true.',
'nbnd': 12},
'electrons': {'diago_full_acc': '.false.', 'conv_thr': 1e-06},
'ions': {},
'cell': {},
'atomic_species': {'Ga': ['1.0', 'Ga_rel.pz-rrkj3.UPF'],
'As': ['1.0', 'As_rel.pz-rrkj3.UPF']},
'atomic_positions': {'type': 'alat',
'values': [['Ga', [0.0, 0.0, 0.0]], ['As', [0.25, 0.25, 0.25]]]},
'kpoints': {'type': 'tpiba_b',
'values': [[0.5, 0.5, 0.5, 20],
[0.0, 0.0, 0.0, 20],
[0.0, 0.0, 1.0, 20],
[0.0, 1.0, 1.0, 20],
[0.0, 0.0, 0.0, 0]]},
'cell_parameters': {},
'file': 'IO_files/gaas_scf.in'}
[60]:
results_gaas_so = code.run(input=inp,run_dir=run_dir,name=bands_prefix,source_dir=os.path.join(run_dir,scf_prefix)+'.save')
delete log file: Pw_bands/gaas_bands_so.log
delete folder: /home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/gaas_bands_so.save
copy source_dir Pw_bands/gaas_scf_so.save in the /home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/gaas_bands_so.save
run command: mpirun -np 4 pw.x -inp gaas_bands_so.in > gaas_bands_so.log
computation gaas_bands_so is running...
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
computation gaas_bands_so ended
Once that the computation is over we can create an instance of BandStructure
[61]:
bands_gaas_so = U.BandStructure.from_Pw(results_gaas_so,hsp,set_gap=1.42)
Apply a scissor of 1.0018908645131264 eV
[62]:
%matplotlib inline
plt.title('GaAs bands with spin-orbit')
plt.ylim(-8,4)
bands_gaas_so.plot(plt,selection=[2,3,4,5,6,7,8,9,10,11])
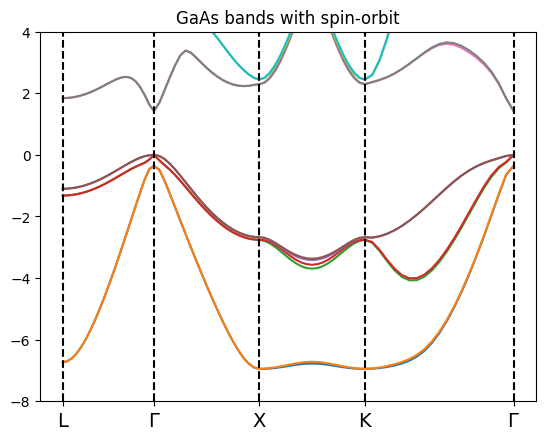
Note that in this case we need to display twice the bands respect to the previous example since each band accomodate only one electron in a specific spin state.
Band structure of Silicon¶
The first step consists in a scf computation
[63]:
scf_prefix = 'si_scf'
bands_prefix = 'si_bands'
[64]:
inp = I.PwInput(file='IO_files/si_scf.in')
inp.set_prefix(scf_prefix)
inp.set_energy_cutoff(60)
inp.set_kpoints(points=[6,6,6])
#inp
[66]:
rr = C.RunRules(mpi=mpi,omp_num_threads=omp)
code = C.QeCalculator(rr)
code.global_options()
Initialize a QuantumESPRESSO calculator with scheduler direct
[66]:
{'scheduler': 'direct',
'mpi': 4,
'omp_num_threads': 1,
'executable': 'pw.x',
'skip': True,
'clean_restart': True,
'dry_run': False,
'wait_end_run': True,
'activate_BeeOND': False,
'verbose': True}
[67]:
code.run(input=inp,run_dir=run_dir,name=scf_prefix)
run command: mpirun -np 4 pw.x -inp si_scf.in > si_scf.log
computation si_scf is running...
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
computation si_scf ended
[67]:
'/home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/si_scf.save/data-file-schema.xml'
Now we perform the bands computation specifying the kpoints on a path.
To define the path we write the coordinates of the high symmetry points (using the tpiba_b type of pw) and we make usage of the function build_kpath
[69]:
hsp = U.Constants.high_sym_fcc
[72]:
klist = C.Tools.build_pw_kpath(hsp['L'],hsp['G'],hsp['X'],hsp['K'],hsp['G'],numstep=20)
klist
[72]:
[[0.5, 0.5, 0.5, 20],
[0.0, 0.0, 0.0, 20],
[0.0, 0.0, 1.0, 20],
[0.0, 1.0, 1.0, 20],
[0.0, 0.0, 0.0, 0]]
[73]:
inp.set_bands(8)
inp.set_prefix(bands_prefix)
inp.set_kpoints(type='tpiba_b',klist=klist)
#inp
[75]:
results_si = code.run(input=inp,run_dir=run_dir,name=bands_prefix,source_dir=os.path.join(run_dir,scf_prefix)+'.save')
copy source_dir Pw_bands/si_scf.save in the /home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/si_bands.save
run command: mpirun -np 4 pw.x -inp si_bands.in > si_bands.log
computation si_bands is running...
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
computation si_bands ended
Once that the computation is over we can create an instance of PwBands
[77]:
bands_si = U.BandStructure.from_Pw(results_si,hsp,set_gap=1.2)
Apply a scissor of 0.6041599954868155 eV
It contains also a plot method that show the band structure
[78]:
%matplotlib inline
plt.title('Si bands')
plt.ylim(-5,4)
bands_si.plot(plt,selection=[1,2,3,4])

Band structure of Graphene¶
[79]:
scf_prefix = 'graphene_scf'
nscf_prefix = 'graphene_nscf'
bands_prefix = 'graphene_bands'
The lattice is oriented with the zig-zag along the y axis. The basis vector are chosen as:
Here \(a\) represents the CC nn distance. The position of the atoms in the lattice is given by:
The lattice constant is given by \(a_{lat} = \sqrt{3}a\).
The position of the high symmetry points of the reciprocal lattice (in units of \(2\pi/a_{alat}\)) are given by:
[84]:
pseudo_pbe = 'C_pbe-20082014.UPF' #pbe
delta = 15 # Angstrom
a0 = 1.42495 # PBE cc equilibrium distance among nearest C atoms
engCutoff = 60 # Ry
numKpoints = 12
import numpy as np
a1 = [a0/2.*3.,a0/2.*np.sqrt(3.),0.]
a2 = [a0/2.*3.,-a0/2.*np.sqrt(3.),0.]
a3 = [0.,0.,delta]
A = [0.,0.,0.]
B = [a0/2.,a0/2.*np.sqrt(3.),0.]
[87]:
inp = I.PwInput()
inp.set_scf()
inp.set_prefix(scf_prefix)
inp.set_pseudo_dir('../pseudos')
inp.add_atom(atom='C',pseudo_name=pseudo_pbe,mass=12.011)
inp.set_atoms_number(2)
inp.set_energy_cutoff(engCutoff)
inp.set_atomic_positions([['C',A],['C',B]],type='angstrom')
inp.set_lattice(ibrav=0,cell_vectors=[a1,a2,a3],cell_units='angstrom')
inp.set_occupations(occupations='smearing',degauss=50.)
inp.set_kpoints(points = [numKpoints,numKpoints,1])
inp
[87]:
{'control': {'calculation': "'scf'",
'verbosity': "'high'",
'prefix': "'graphene_scf'",
'outdir': "'./'",
'pseudo_dir': "'../pseudos'"},
'system': {'force_symmorphic': '.false.',
'ntyp': '1',
'nat': '2',
'ecutwfc': 60,
'ibrav': 0,
'occupations': "'smearing'",
'smearing': "'fermi-dirac'",
'degauss': 0.00367493225078649},
'electrons': {'diago_full_acc': '.false.', 'conv_thr': 1e-08},
'ions': {},
'cell': {},
'atomic_species': {'C': [12.011, 'C_pbe-20082014.UPF']},
'atomic_positions': {'type': 'angstrom',
'values': [['C', [0.0, 0.0, 0.0]],
['C', [0.712475, 1.2340428991226358, 0.0]]]},
'kpoints': {'type': 'automatic', 'values': ([12, 12, 1], [0.0, 0.0, 0.0])},
'cell_parameters': {'type': 'angstrom',
'values': [[2.137425, 1.2340428991226358, 0.0],
[2.137425, -1.2340428991226358, 0.0],
[0.0, 0.0, 15]]}}
[88]:
rr = C.RunRules(mpi=mpi,omp_num_threads=omp)
code = C.QeCalculator(rr)
#code.global_options()
Initialize a QuantumESPRESSO calculator with scheduler direct
[89]:
code.run(input=inp,run_dir=run_dir,name=scf_prefix)
delete log file: Pw_bands/graphene_scf.log
run command: mpirun -np 4 pw.x -inp graphene_scf.in > graphene_scf.log
computation graphene_scf is running...
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
computation graphene_scf ended
[89]:
'/home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/graphene_scf.save/data-file-schema.xml'
We perfrom also a nscf computation on a regular grid of kpoint. Results of this computation are used by the other tutorials of the package
[91]:
inp.set_nscf(8)
inp.set_prefix(nscf_prefix)
#inp
[93]:
code.run(input=inp,run_dir=run_dir,name=nscf_prefix,source_dir=os.path.join(run_dir,scf_prefix)+'.save')
copy source_dir Pw_bands/graphene_scf.save in the /home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/graphene_nscf.save
run command: mpirun -np 4 pw.x -inp graphene_nscf.in > graphene_nscf.log
computation graphene_nscf is running...
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
computation graphene_nscf ended
[93]:
'/home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/graphene_nscf.save/data-file-schema.xml'
Now we perform the bands computation specifying the kpoints on a path.
To define the path we write the coordinates of the high symmetry points (using the tpiba_b type of pw) and we make usage of the function build_kpath
[94]:
G = [0.,0.,0.]
M = [1./np.sqrt(3.),0.,0.]
K = [1./np.sqrt(3.),1./3.,0.]
# useful to label the high-sym point on the path
high_sym = {'G':G,'K':K,'M':M}
[96]:
klist = C.Tools.build_pw_kpath(G,M,K,G,numstep=30)
#klist
[97]:
inp.set_bands(8)
inp.set_prefix(bands_prefix)
inp.set_kpoints(type='tpiba_b',klist=klist)
#inp
[98]:
results_gra = code.run(input=inp,run_dir=run_dir,name=bands_prefix,source_dir=os.path.join(run_dir,scf_prefix)+'.save')
copy source_dir Pw_bands/graphene_scf.save in the /home/marco/Applications/MPPI/sphinx_source/tutorials/Pw_bands/graphene_bands.save
run command: mpirun -np 4 pw.x -inp graphene_bands.in > graphene_bands.log
computation graphene_bands is running...
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
Note: The following floating-point exceptions are signalling: IEEE_DENORMAL
computation graphene_bands ended
Once that the computation is over we can create an instance of PwBands
[99]:
bands_gra = U.BandStructure.from_Pw(results_gra,high_sym)
It contains also a plot method that show the band structure
[100]:
bands_gra.get_high_sym_positions()
[100]:
(['$\\Gamma$', '$\\Gamma$', 'K', 'M'],
[0.0, 1.5773502657018026, 0.9106835999999987, 0.57735027])
[101]:
%matplotlib inline
plt.title('Graphene bands',size=14)
#plt.ylim(-6,8)
bands_gra.plot(plt) #,selection=[0,1,2,3,4,5,6,7]
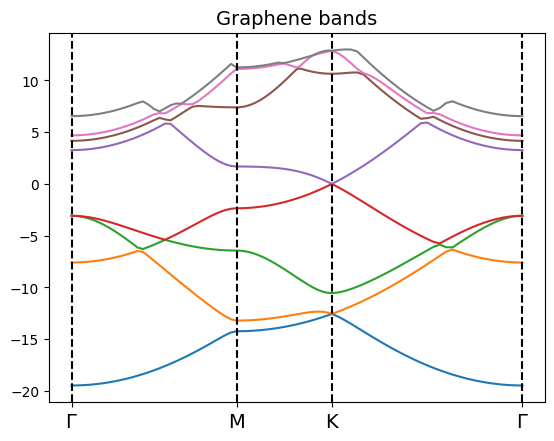
Analysis of the band structure with ypp¶
[4]:
run_dir = 'Ypp_bands'
Now we analyze the band structure using the post processing tools of ypp.
Band structure of GaAs with spin-orbit coupling¶
As an example we consider the GaAs band structure with spin-orbit coupling.
We perform a ypp -s b computation starting from the .save of an nscf pw computation on a regular grid
[5]:
source_dir = C.Tools.make_p2y('Pw_bands/gaas_bands_so.save')
source_dir
SAVE folder Pw_bands/gaas_bands_so.save/SAVE already present. No operations performed.
[5]:
'Pw_bands/gaas_bands_so.save/SAVE'
[107]:
C.Tools.init_yambo_run_dir(source_dir,run_dir)
Create folder path Ypp_bands
Create a symlink of Pw_bands/gaas_bands_so.save/SAVE in Ypp_bands
Build the r_setup in the run_dir path Ypp_bands
[6]:
rr = C.RunRules(mpi=mpi,omp_num_threads=1)
code = C.YamboCalculator(rr,executable='ypp')
code.global_options()
Initialize a Yambo calculator with scheduler direct
[6]:
{'scheduler': 'direct',
'mpi': 4,
'omp_num_threads': 1,
'executable': 'ypp',
'skip': True,
'clean_restart': True,
'dry_run': False,
'wait_end_run': True,
'activate_BeeOND': False,
'verbose': True,
'fatlog': False}
[48]:
inp = I.YamboInput(args='ypp -s b -V qp',folder=run_dir,filename='ypp.in')
inp
[48]:
{'args': 'ypp -s b -V qp',
'folder': 'Ypp_bands',
'filename': 'ypp.in',
'arguments': [],
'variables': {'INTERP_Shell_Fac': [20.0, ''],
'INTERP_NofNN': [1.0, ''],
'OutputAlat': [0.0, ''],
'BANDS_steps': [10.0, ''],
'GfnQP_INTERP_NN': [1.0, ''],
'GfnQP_INTERP_shells': [20.0, ''],
'GfnQP_Wv_E': [0.0, 'eV'],
'GfnQP_Wv_dos': [0.0, 'eV'],
'GfnQP_Wc_E': [0.0, 'eV'],
'GfnQP_Wc_dos': [0.0, 'eV'],
'PROJECT_mode': 'none',
'INTERP_mode': 'NN',
'cooIn': 'rlu',
'cooOut': 'rlu',
'CIRCUIT_E_DB_path': 'none',
'GfnQPdb': 'none',
'GfnQP_DbGd_INTERP_mode': 'NN',
'GfnQP_Z': [(1+0j), ''],
'BANDS_bands': [[1, 12], ''],
'GfnQP_E': [[0.0, 1.0, 1.0], ''],
'GfnQP_Wv': [[0.0, 0.0, 0.0], ''],
'GfnQP_Wc': [[0.0, 0.0, 0.0], '']}}
Set the input parameter to perform the band computation along a path
[49]:
# set the coordinate of the high-sym points (use crystal coordinates) to be consistent with th
# rlu choice in the cooIn and cooOut variables in the ypp input
G = [0.,0.,0.]
X = [0.,0.5,0.5]
L = [0.5,0.5,0.5]
K = [3./8.,3/4.,3/8.]
W = [1./4.,3./4.,1./2.]
# useful to label the high-sym point on the path
high_sym = {'X':X,'L':L,'G':G,'K':K,'W':W}
# set the path (use the same path of the pw computation)
path = [L,G,X,K,G]
# set the number of intermediate points between two high-sym ones
bands_step = 20
[51]:
# scissor
# inp['variables']['GfnQP_E'] = [1.0,1.0,1.0]
# band structure
inp.set_scalar_variables(INTERP_mode='NN')
inp.set_array_variables(BANDS_steps=bands_step,BANDS_kpts=path,INTERP_Shell_Fac=20)
#inp
[52]:
results_ypp = code.run(run_dir=run_dir,input=inp,name='gaas_bands_so',skip=False)
results_ypp
delete folder: Ypp_bands/gaas_bands_so
run command: mpirun -np 4 ypp -F gaas_bands_so.in -J gaas_bands_so -C gaas_bands_so
computation gaas_bands_so is running...
computation gaas_bands_so ended
Run performed in 05s
[52]:
{'output': {'magnetization_z_interpolated': 'Ypp_bands/gaas_bands_so/o-gaas_bands_so.magnetization_z_interpolated',
'spin_factors_DN_interpolated': 'Ypp_bands/gaas_bands_so/o-gaas_bands_so.spin_factors_DN_interpolated',
'magnetization_y_interpolated': 'Ypp_bands/gaas_bands_so/o-gaas_bands_so.magnetization_y_interpolated',
'spin_factors_UP_interpolated': 'Ypp_bands/gaas_bands_so/o-gaas_bands_so.spin_factors_UP_interpolated',
'bands_interpolated': 'Ypp_bands/gaas_bands_so/o-gaas_bands_so.bands_interpolated',
'magnetization_x_interpolated': 'Ypp_bands/gaas_bands_so/o-gaas_bands_so.magnetization_x_interpolated'},
'report': 'Ypp_bands/gaas_bands_so/r-gaas_bands_so_electrons_bnds',
'dft': 'Ypp_bands/SAVE/ns.db1'}
Once that the computation is over we can create an instance of PwBands
[53]:
U.BandStructure.from_Ypp?
Signature:
U.BandStructure.from_Ypp(
results,
high_sym_points=None,
suffix='bands_interpolated',
)
Docstring:
Initialize the BandStructure class from the data dictionay of the result of a Ypp postprocessing.
The class make usage of the YamboParser of this package.
Args:
results (:py:class:`dict`) : dictionary with the output of a Ypp computation
high_sym_points(:py:class:`dict`) : dictionary with name and coordinates of the
high_sym_points of the path
suffix (string) : specifies the suffix of the o- file use to build the bands
File: ~/Applications/MPPI/mppi/Utilities/BandStructure.py
Type: method
[54]:
bands_ypp = U.BandStructure.from_Ypp(results_ypp,high_sym)
[55]:
%matplotlib inline
plt.title('GaAs band from Ypp with spin-orbit',size=14)
plt.ylim(-8,3)
bands_ypp.plot(plt,selection=[2,3,4,5,6,7,8,9,10,11])
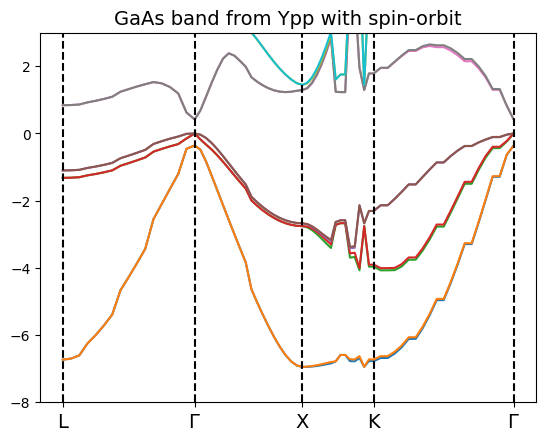
Note that the NN interpolation method has been used, since the BOLTZ (which is the right one for the band structure evaluation) gives an interpolation error at run time, to be fixed!
The design of the plot method of the class allows us to plot bands coming from different calculations in the same plot.
For instance, we can show the effect of the spin-orbit on the valence band of GaAs
[61]:
bands_gaas.plot(plt,selection= [1,3],label='no_so',show_vertical_lines=False)
bands_gaas_so.plot(plt,selection = [2,6],ls='--',label='so',show_vertical_lines=False)
plt.ylim(-4,0.5)
plt.xlim(0,2)
plt.legend()
[61]:
<matplotlib.legend.Legend at 0x7f8db8b9f278>

The shift of the split-off band is visible.